Gene-Oriented Ortholog Database
Origion
Orthology is a widely used concept in comparative and evolutionary genomics. In addition to prokaryotic orthology, delineating eukaryotic orthology has provided insight into the evolution of higher organisms. Unlike prokaryotes, alternative splicing (AS) effect of genes, a main way to increasing the biological complexity, has hampered the orthology assignments of eukaryotes, especially for higher eukaryotes. Therefore, existing databases contain ambiguous and incomplete eukaryotic ortholog relationships and possibly misclassify AS isoforms as in-paralogs, which are duplicated genes that arise following speciation.
Ortholog database
Gene-Oriented Ortholog Database (GOOD) clusters all alternative splicing (AS)-derived isoforms associated with each transcription region (gene) prior to ortholog delineation and then derives processed transcription units (PTUs) from the union of all the AS transcripts of one gene (Figure 1).
Figure 1.
![]()
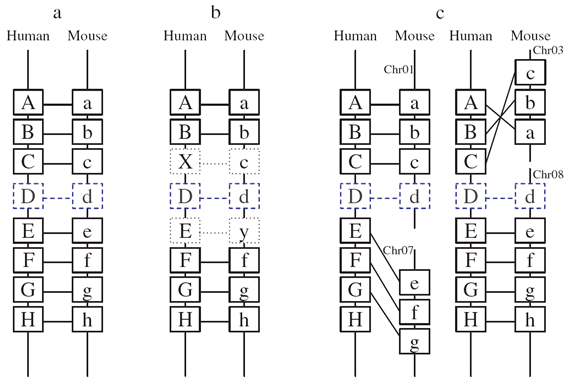
GOOD is mainly based on the Best Reciprocal Hits (BRHs) of PTUs as anchor pairs, putative orthologs, ( Figure 2, boxes outlined in black). The remaining aligned PTU pairs were assigned as potential pairs ( Figure 2, boxes outlined in blue). Take human and mouse for instance, we map anchor pairs back to their genomic locations, and discovered a pre-existing syntenic relationship between the human and mouse genomes (Figure 2). Then we further examined the potential pairs individually and added those pairs that fit into the syntenic anchor structure (Figure 2, D-d pair). Three criteria were applied to determine fit ( Figure 2, a, b, and c). For detail information, please refer to "Designating eukaryotic orthology via processed transcription units".
Figure 2.
Graphical presentation of Gene Ontology (GO) terms
GOOD takes in functional annotations from the Gene Ontology (GO) database. GO terms of the same orthologous gene are displayed with respect to three structured controlled vocabularies (ontologies), biological processes, cellular components, and molecular functions. Cooperating with functional annotations from GO, GOOD serves as a functional comparison platform for orthologous loci. Simultaneous presentation of two desired orthologous genomic loci allows researchers to rapidly identify differences in both alternative splicing events and gene functions between orthologs.
Graphs of all GO terms are generated using the following three steps. Step 1: For the queried GO term, all possible paths to the root are found. Step 2: The longest path is assigned as the center path and plotted in the center of the graph. Step 3: The longest and most common path to the center path is selected from the remaining paths and is included on the graph next to the existing paths. Step 4: Reiterate Step 3 until all possible paths are plotted.
Using graphs, GOOD displays not only the terminal nodes of GO annotations but also all possible paths from the annotated terms to the root. This graphical presentation promotes elucidation of functions’ hierarchical structure. For functional annotation from GO, please visit the Gene Ontology.